VAPEX: Virus And Phage EXplorer
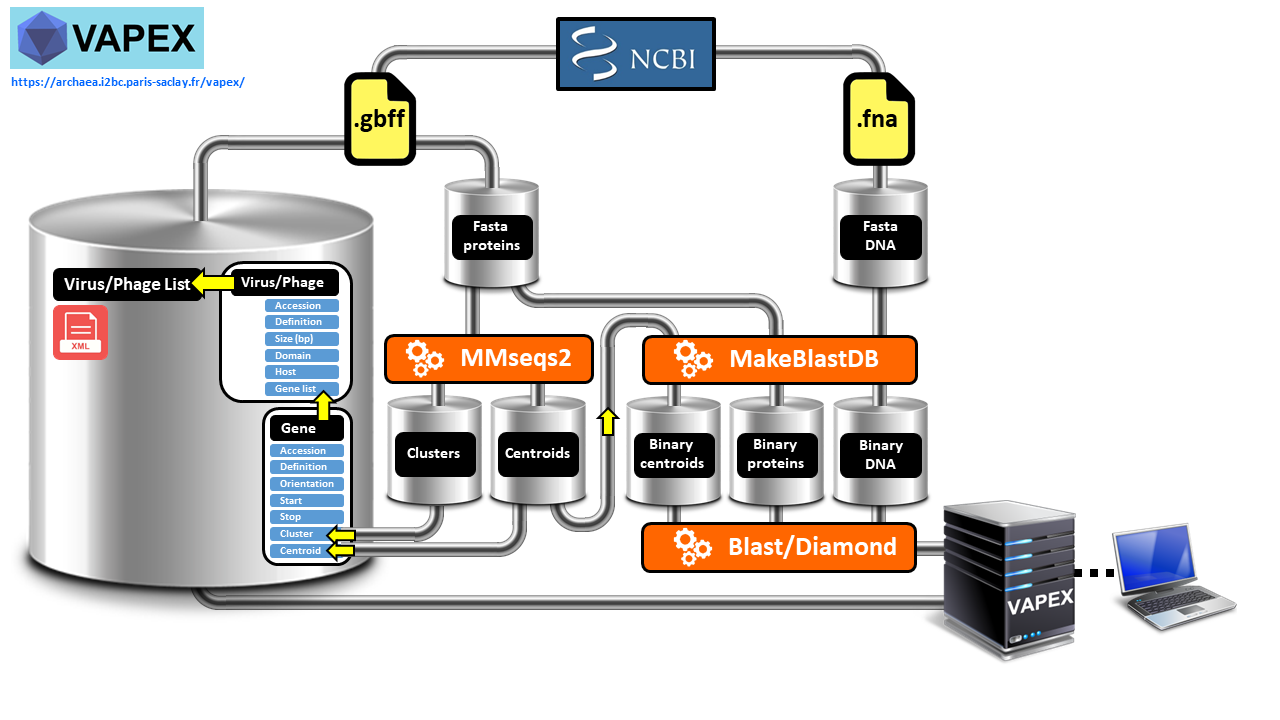
We have developed VAPEX (Virus And Phage EXplorer), a web server which is supported by a database and features a user-friendly web interface. This tool enables users to easily perform various genomic analysis queries on all natural viruses and phages that have been fully sequenced and are listed in the NCBI compendium. VAPEX therefore excels in producing visual depictions of fully resolved synteny maps, which is one of its key strengths. VAPEX has the ability to exhibit a vast array of orthologous gene classes simultaneously through the use of symbolic representation. Additionally, VAPEX can fully analyze user-submitted viral and phage genomes, including those that have not yet been annotated.
SYNTTAX: Prokaryotic Synteny & Taxonomy Explorer
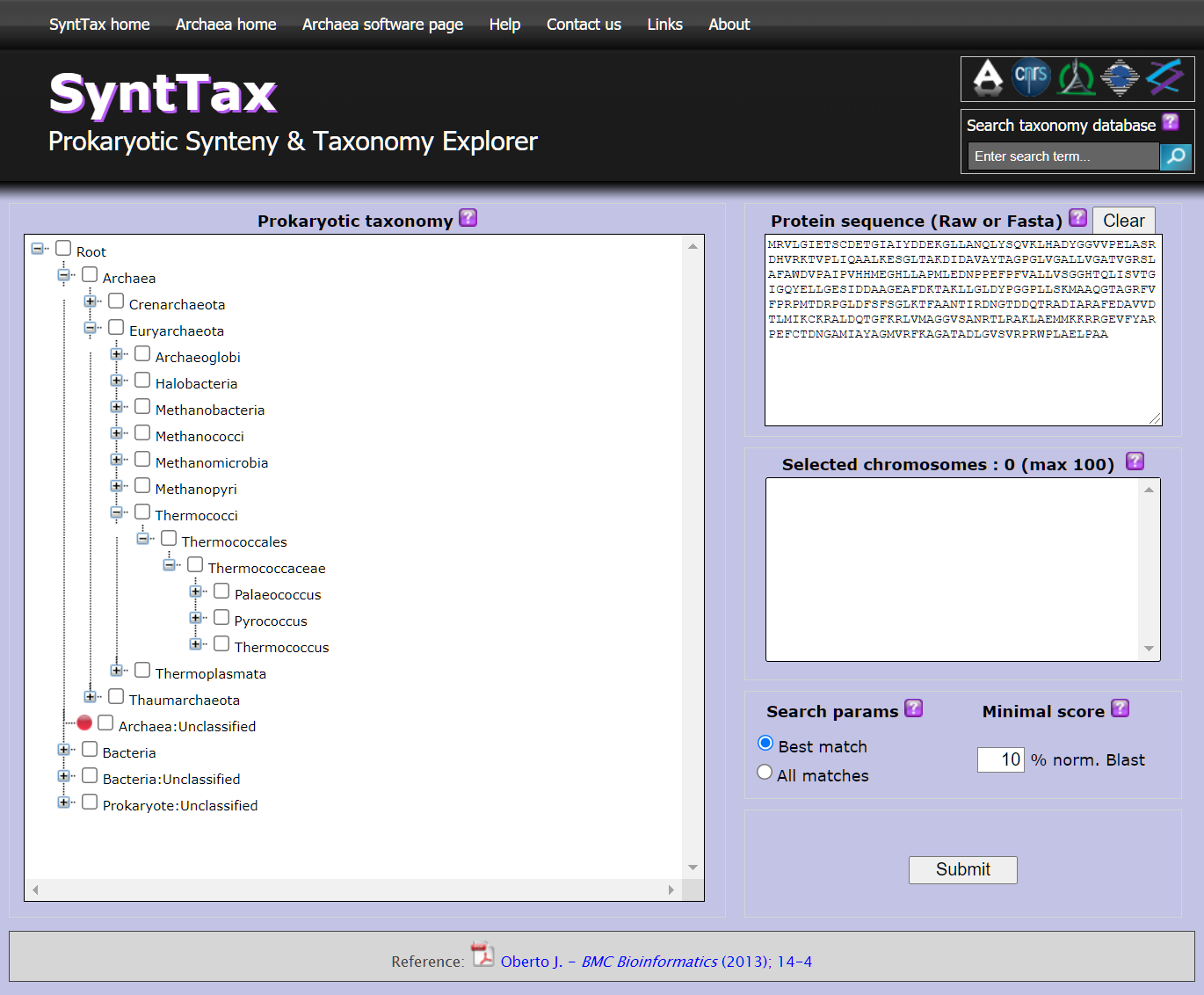
The study of the conservation of gene order or synteny constitutes a powerful methodology to assess the orthology of genomic regions and to predict functional relationships between genes. The exponential growth of microbial genomic databases is expected to improve synteny predictions significantly. Paradoxically, this genomic data plethora, without information on organisms relatedness, could impair the performance of synteny analysis programs. In this work, I present SyntTax, a synteny web service designed to take full advantage of the large amount or archaeal and bacterial genomes by linking them through taxonomic relationships. SyntTax incorporates a full hierarchical taxonomic tree allowing intuitive access to all completely sequenced prokaryotes.

WASPS: Web-Assited Symbolic Plasmid Synteny
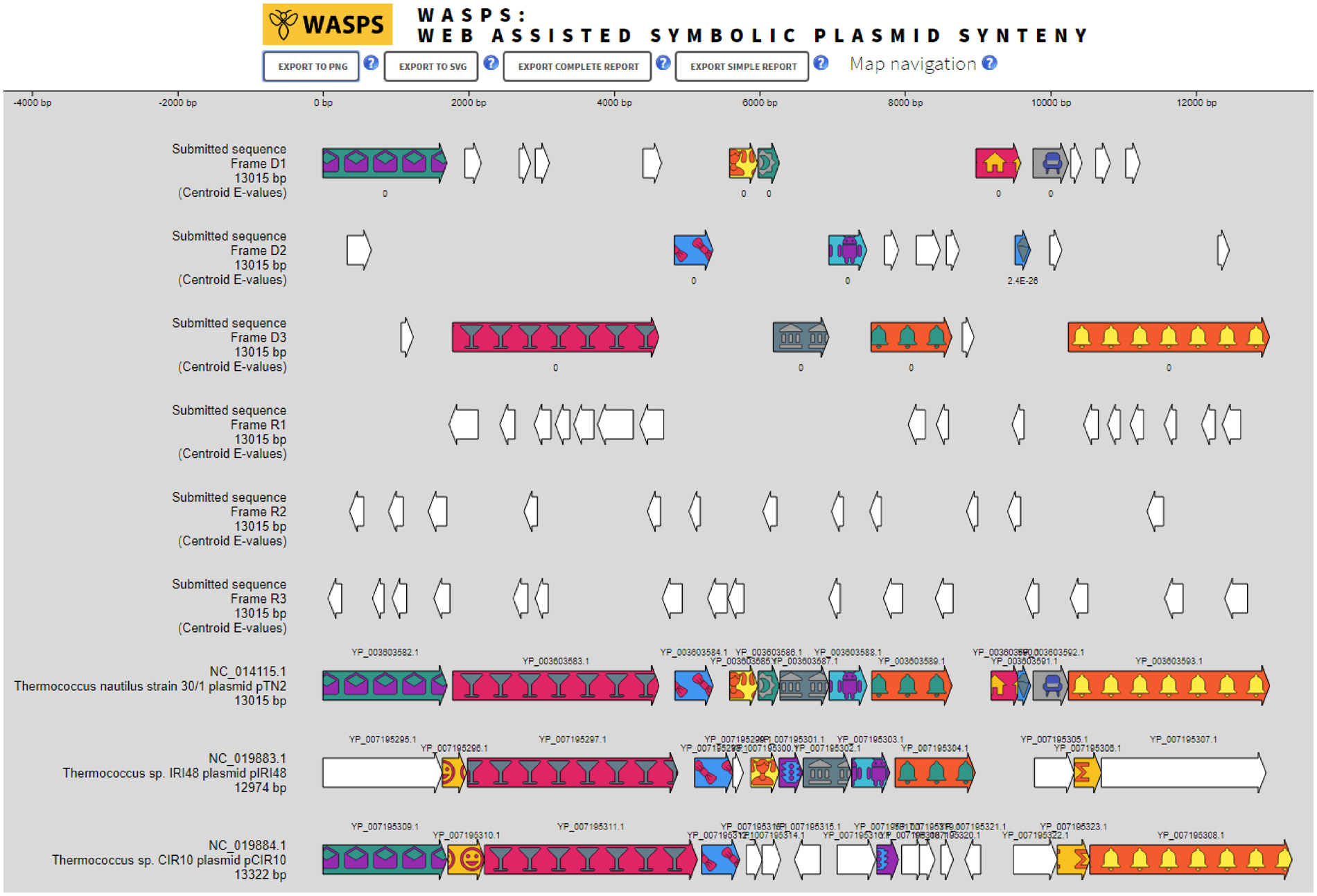
Comparative plasmid genome analyses require complex tools, the manipulation of large numbers of sequences and constitute a daunting task for the wet bench experimentalist. Dedicated plasmid databases are sparse, only comprise bacterial plasmids and provide exclusively access to sequence similarity searches. We have developed Web-Assisted Symbolic Plasmid Synteny (WASPS), a web service granting protein and DNA sequence similarity searches against a database comprising all completely sequenced natural plasmids from bacterial, archaeal and eukaryal origin. This database pre-calculates orthologous protein clustering and enables WASPS to generate fully resolved plasmid synteny maps in real time using internal and user-provided DNA sequences.

BAGET 2.0: Bacterial and Archaeal Gene Exploration Tool
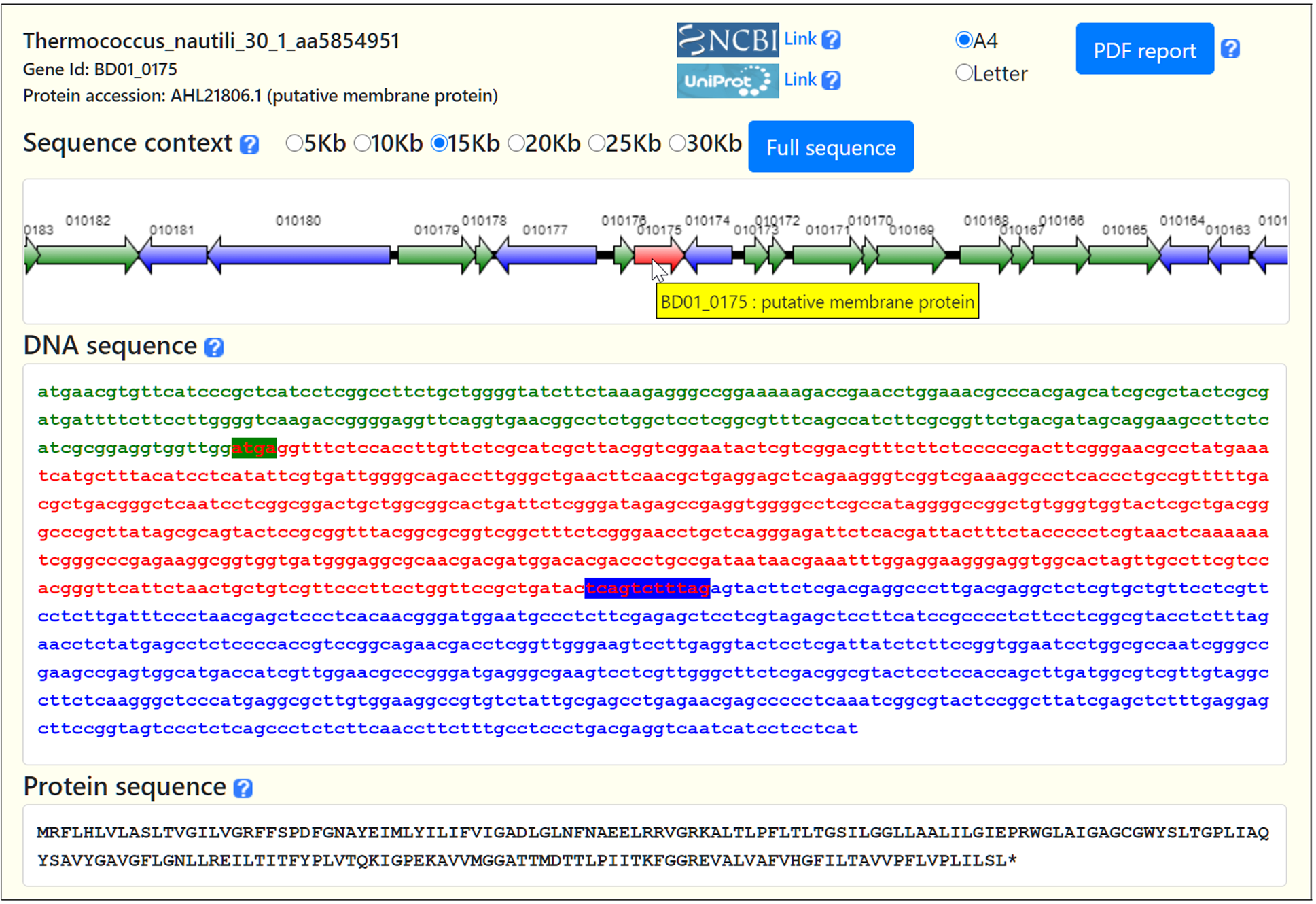
The retrieval of a single gene sequence and context from completely sequenced bacterial and archaeal genomes constitutes an intimidating task for the wet bench biologist. Existing web-based genome browsers are either too complex for routine use or only provide a subset of the available prokaryotic genomes. We have developed BAGET 2.0 (Bacterial and Archaeal Gene Exploration Tool), an updated web service granting access in just three mouse clicks to the sequence and synteny of any gene from completely sequenced bacteria and archaea. User-provided annotated genomes can be processed as well. BAGET 2.0 relies on a local database updated on a daily basis.

FITBAR: Fast Investigation Tool for Bacterial and Archaeal Regulons
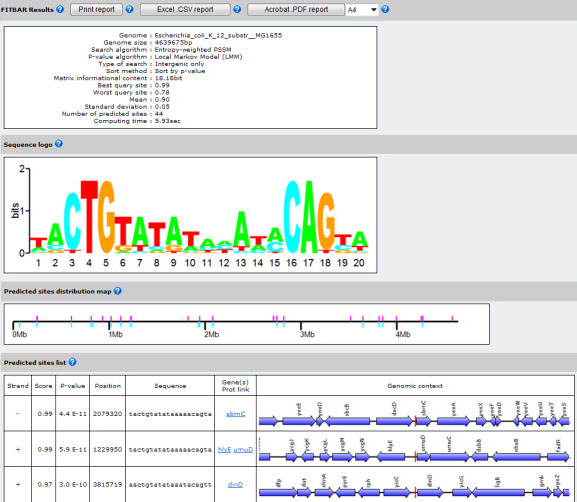
The binding of regulatory proteins to their specific DNA targets determines the accurate expression of the neighboring genes. The in silico prediction of new binding sites in completely sequenced genomes is a key aspect in the deeper understanding of gene regulatory networks. Several algorithms have been described to discriminate against false-positives in the prediction of new binding targets; however none of them has been implemented so far to assist the detection of binding sites at the genomic scale. FITBAR (Fast Investigation Tool for Bacterial and Archaeal Regulons) is a web service designed to identify new protein binding sites on fully sequenced prokaryotic genomes. The Local Markov Model and the Compound Importance Sampling algorithms have been implemented to compute the P-value of newly discovered binding sites. In addition, FITBAR provides two optimized genomic scanning algorithms using either log-odds or entropy-weighted position-specific scoring matrices.

Sponsors



