
| This is the FITBAR help page |
| Abstract |
The binding of regulatory proteins to their specific DNA targets determines the accurate
expression of the neighboring genes. The in silico prediction of new binding sites in
completely sequenced genomes is a key aspect in the deeper understanding of gene regulatory
networks. Several algorithms have been described to discriminate against false-positives
in the prediction of new binding targets; however none of them has been implemented so
far to assist the detection of binding sites at the genomic scale.
FITBAR (Fast Investigation Tool for Bacterial and Archaeal Regulons) is a web service designed to identify new protein binding sites on fully sequenced prokaryotic genomes. This tool consists in a workbench where the significance of the predictions can be compared using different statistical methods, a feature not found in existing resources. The Local Markov Model and the Compound Importance Sampling algorithms have been implemented to compute the P-value of newly discovered binding sites. In addition, FITBAR provides two optimized scanning algorithms using either log-odds or entropy-weighted position-specific scoring matrices. Other significant features include the production of a detailed genomic context map for each detected binding site and the export of the search results in spreadsheet and portable document formats. FITBAR discovery of a high affinity Escherichia coli NagC binding site was validated experimentally in vitro as well as in vivo and published.
FITBAR was developed in order to allow fast, accurate and statistically robust predictions of prokaryotic regulons. This feature constitutes the main advantage of this web tool over other matrix search programs and does not impair its performance. The web service is available at http://archaea.u-psud.fr/fitbar.
FITBAR (Fast Investigation Tool for Bacterial and Archaeal Regulons) is a web service designed to identify new protein binding sites on fully sequenced prokaryotic genomes. This tool consists in a workbench where the significance of the predictions can be compared using different statistical methods, a feature not found in existing resources. The Local Markov Model and the Compound Importance Sampling algorithms have been implemented to compute the P-value of newly discovered binding sites. In addition, FITBAR provides two optimized scanning algorithms using either log-odds or entropy-weighted position-specific scoring matrices. Other significant features include the production of a detailed genomic context map for each detected binding site and the export of the search results in spreadsheet and portable document formats. FITBAR discovery of a high affinity Escherichia coli NagC binding site was validated experimentally in vitro as well as in vivo and published.
FITBAR was developed in order to allow fast, accurate and statistically robust predictions of prokaryotic regulons. This feature constitutes the main advantage of this web tool over other matrix search programs and does not impair its performance. The web service is available at http://archaea.u-psud.fr/fitbar.
| How does it work? |
FITBAR proceeds in four main steps.
1. FITBAR starts by reading the user-selected binding site matrix, the selected chromosome sequence with associated gene definitions and the chromosome-specific mono, di, tri and tetranucleotide frequencies are read from the database.
2. The position-specific scoring matrix (PSSM) also called position-specific weight matrix (PSWM) is generated at this step from the input consensus sites using algorithms derived from the information theory. It takes into account the background distribution of nucleotides of the selected chromosome.
3. At this stage, the chromosome scanning will proceed on both DNA strands simultaneously. It can take from a few seconds up to one minute depending on the selected chromosome length, the informational content of the consensus and the web server load.
4. If the search is successful, the results are displayed on a new page.
1. FITBAR starts by reading the user-selected binding site matrix, the selected chromosome sequence with associated gene definitions and the chromosome-specific mono, di, tri and tetranucleotide frequencies are read from the database.
2. The position-specific scoring matrix (PSSM) also called position-specific weight matrix (PSWM) is generated at this step from the input consensus sites using algorithms derived from the information theory. It takes into account the background distribution of nucleotides of the selected chromosome.
3. At this stage, the chromosome scanning will proceed on both DNA strands simultaneously. It can take from a few seconds up to one minute depending on the selected chromosome length, the informational content of the consensus and the web server load.
4. If the search is successful, the results are displayed on a new page.
| Select archaeal or bacterial chromosome |
Archaeal and bacterial chromosomes are listed by organism name and can be selected here. Multiple
chromosomes belonging to the same organism are listed with the C1, C2, C3… suffices, by decreasing
size. Chromosomes are selected and searched individually.
| Select DNA binding site matrix from the database |
FITBAR provides a choice of over 200 prokaryotic binding site matrices collected from
Harvard
University and
RegTransBase.
Each consensus can be loaded, viewed and/or edited manually. The consensus site format
is standard and discussed below.
| Select matrix search algorithm |
FITBAR provides a choice of two matrix search algorithms.
They are knows as "log-odds PSSM" and
"entropy-weighted PSSM", respectively.
a) Log-odds PSSM (Durbin et al.,1998)
For each position i and for each base x = A, C, G, or T, a log odds is calculated as follows:
where mi is the probability for observing base x at position i from the
nucleotide distribution matrix, and qx is the probability of observing base x under
a random model.
In our specific case, this probablity is computed from the genomic sequence and corresponds to the true nucleotide
frequency.
b) Entropy-weighted PSSM (Quandt et al., 1995)
The values in the PSSM are derived by weighting the counts in the nucleotide distribution matrix at each position using a entropy-related information measure:
where mi is the probability for observing base x at position i from the nucleotide
distribution matrix.
a) Log-odds PSSM (Durbin et al.,1998)
For each position i and for each base x = A, C, G, or T, a log odds is calculated as follows:
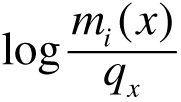 |
b) Entropy-weighted PSSM (Quandt et al., 1995)
The values in the PSSM are derived by weighting the counts in the nucleotide distribution matrix at each position using a entropy-related information measure:
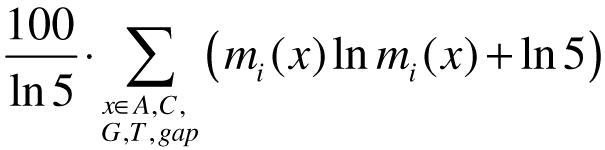 |
| Select p-value algorithm |
FITBAR provides a choice of two p-value calculation alogrithms. The first algorithm is known as the Compound Importance
Sampling (CIS) and was described by Barash et al. (2005).
The second algorithm is based on the Local Markov Method (LMM) and was described by
Huang et al. (2004).
| Select chromosomal search mode |
Two search methods are provided: "Intergenic only" (selected by default) and
"whole genome".
The "Intergenic only" search ignores protein encoding regions and concentrates on non-translated
regions. The "Whole chromosome" search will scan the entire chromosome, regardless of genetic
features.
| Select sort method |
Three binding sites list sorting methods are available "Sort
by p-value" (selected by default), "Sort by score" and
"Sort by position". Self explanatory.
| Enter user-defined matrix |
The binding site matrix can be loaded from the local database (see above). Alternatively, the list of
input binding sites can be added manually or by copy/paste from a local text file directly on the
web page. Mixed case site sequences are accepted. Lines starting with ‘>’ are considered as
comments and not as binding sites. Important: (i) the input binding sites need to be properly aligned;
(ii) all input sites need to be of equal length and (iii) no gaps are allowed. Valid input file example:
|
>E. coli LexA binding sites } comment line : ignored >this is a comment line } comment line : ignored aactgtatataaatacagtt tattggctgtttatacagta tcctgttaatccatacagca >acctgtataaataaccagta } commented out line : ignored tgctgtatatactcacagca aactgtatatacacccaggg |
| FITBAR Results |
If the search is successful, FITBAR will display four bodies of information:
1. Genaral Statistics. Information concerning the selected chromosome is presented here such as organism name, chromosome length and nucleotide distribution. In addition, consensus site statistics are shown: mean score, standard deviation and worst score. The number of new sites found on the chromosome is indicated as well.
2. Sequence logo.
3. Predicted sites distribution map.
4. Predicted sites list.
1. Genaral Statistics. Information concerning the selected chromosome is presented here such as organism name, chromosome length and nucleotide distribution. In addition, consensus site statistics are shown: mean score, standard deviation and worst score. The number of new sites found on the chromosome is indicated as well.
2. Sequence logo.
3. Predicted sites distribution map.
4. Predicted sites list.
| Sequence logo |
This handy representation of a consensus motif was developed originally by
Tom D. Schneider and permits to visually
evaluate the quality (information content) of a DNA binding site consensus.
| Predicted sites distribution map |
This graphic representation of the newly discovered sites on the chromosome permits to evaluate their
distribution, on both DNA strands.
| Predicted sites list |
| Binding site p-value |
The p-value is a commonly accepted measure for the statistical significance of a result. In this particular
case, if 100 predicted binding sites obtain a p-value of 0.01, the prediction would be
exact for 99 sites
and coincidental for one site.
| Print FITBAR Report |
This feature allows the user to print FITBAR results on paper.
This option is for useful for casual data printing;
production-quality printing is better achieved with the .PDF
export feature (see below) Please be environment-friendly and use
moderately: FITBAR reports can be quite extensive and entire forests could be wasted.
| Export FITBAR Excel .CSV Report |
This feature allows the user to save FITBAR results as a local
Excel .CSV file, compatible with all spreadsheet
or word processor programs, for storage or further use. The Excel report contains all the results, with
the exception of the sequence logo, the predicted sites distribution map and the genomic context maps.
This report is generated "on the fly" from system memory: no
user data is ever saved on the server, at any moment.
| Export FITBAR Acrobat .PDF Report |
This feature allows the user to save FITBAR results as a local
Acrobat .PDF file, compatible with Adobe Acrobat Reader on all systems
for printing, storage or further use. The Acrobat report contains all the results, including
the sequence logo, the predicted sites distribution map and
all genomic context maps. This feature allows
device-independent, high-resolution printing in A4 and
Letter formats. This report is generated "on the fly" from
system memory: no user data is ever saved on the server, at
any moment.
| Genomic database updates |
All the FITBAR genomic databases are stored on the server. Their update is fully automated and occurs daily at 07:00 GMT/Zulu.
| Useful links |
•
Compound importance sampling
[Medline]
• Sequence logos [Medline]
• Predict Regulon Server [Medline]
• TESS : Transcription Element Search System [Ref.]
• PRODORIC Virtual Footprint [Medline]
• RSA Tools [Medline]
• BAGET: a web server for the effortless retrieval of prokaryotic gene context and sequence [Medline]
• Harvard University DNA binding matrix collection
• RegTansBase DNA binding matrix collection
• National Center for Biotechnology Information (NCBI)
• ARCHAEA software page
• Sequence logos [Medline]
• Predict Regulon Server [Medline]
• TESS : Transcription Element Search System [Ref.]
• PRODORIC Virtual Footprint [Medline]
• RSA Tools [Medline]
• BAGET: a web server for the effortless retrieval of prokaryotic gene context and sequence [Medline]
• Harvard University DNA binding matrix collection
• RegTansBase DNA binding matrix collection
• National Center for Biotechnology Information (NCBI)
• ARCHAEA software page

Help file last updated 2010 Jun 17.