SyntTax
SyntTax
Prokaryotic Synteny & Taxonomy Explorer
| This is the SyntTax help page |
| Abstract |
Background: The study of the conservation of gene order or synteny constitutes a powerful methodology to assess the orthology of genomic regions and to predict functional relationships between genes. The exponential growth of microbial genomic databases is expected to improve synteny predictions significantly. Paradoxically, this genomic data plethora, without information on organisms relatedness, could impair the performance of synteny analysis programs.
Results: In this work, I present SyntTax, a synteny web service designed to take full advantage of the large amount or archaeal and bacterial genomes by linking them through taxonomic relationships. SyntTax proposes the biologist a full hierarchical taxonomic tree allowing intuitive access to all completely sequenced prokaryotes. Single or multiple organisms can be chosen on the basis of their lineage by selecting the corresponding rank nodes in the tree. The synteny methodology is built upon our previously described Absynte algorithm with several additional improvements.
Conclusions: SyntTax aims to produce robust syntenies by providing a prompt access to the taxonomic relationships connecting all completely sequenced microbial genomes. The reduction in redundancy offered by lineage selection presents the benefit of increasing accuracy while reducing computation time. This web tool was used to resolve successfully several complex syntenies described in the literature. In addition, particular features of SyntTax permitted to propose an additional function for the E. coli YgjD multiprotein complex by analyzing the clustering evolution of alternative gene fusions. This complex is predicted to link the processes of cell wall synthesis and tRNA modification. The web service is available at http://archaea.u-psud.fr/SyntTax.
Reference: submitted for review
Results: In this work, I present SyntTax, a synteny web service designed to take full advantage of the large amount or archaeal and bacterial genomes by linking them through taxonomic relationships. SyntTax proposes the biologist a full hierarchical taxonomic tree allowing intuitive access to all completely sequenced prokaryotes. Single or multiple organisms can be chosen on the basis of their lineage by selecting the corresponding rank nodes in the tree. The synteny methodology is built upon our previously described Absynte algorithm with several additional improvements.
Conclusions: SyntTax aims to produce robust syntenies by providing a prompt access to the taxonomic relationships connecting all completely sequenced microbial genomes. The reduction in redundancy offered by lineage selection presents the benefit of increasing accuracy while reducing computation time. This web tool was used to resolve successfully several complex syntenies described in the literature. In addition, particular features of SyntTax permitted to propose an additional function for the E. coli YgjD multiprotein complex by analyzing the clustering evolution of alternative gene fusions. This complex is predicted to link the processes of cell wall synthesis and tRNA modification. The web service is available at http://archaea.u-psud.fr/SyntTax.
Reference: submitted for review
| SyntTax workflow |
The user is provided with an exhaustive taxonomic tree
covering all the completely sequenced archaeal and bacterial organisms
available at the
NCBI.
Single or multiple organisms can be selected intuitively by
chosing the appropriate taxonomic ranks.
Upon submission of a protein sequence, SyntTax computes the local genomic context (or synteny)
of the corresponding homologous gene originating from a user-selected list of
fully sequenced sequenced archaeal or bacterial genomes.
SyntTax displays local genomic maps drawn to scale and with
a consistent color code and to allow immediate comparative
visual analysis of the gene order conservation in the
selected organisms. SyntTax queries are computationally
intensive tasks; several solutions have been developed to
increase performance. The SyntTax workflow is executed
locally on the server and consists of seven major steps:
Step 1. The protein sequence is matched against itself using BLASTP and the resulting bit score is used as the reference score (100%).
Step 2. The query protein is matched against the selected chromosomes translated in the six frames using the TBLASTN algorithm.
Step 3. The resulting scores are normalized according to the reference score determined above. Only the scores above a user-selected threshold are retained (default and minimal value of 10%). The user will also determine if only one score per chromosome or all scores are retained. The chromosomes are then ranked by decreasing scores.
Step 4. For each positive scoring chromosome, SyntTax pulls out a DNA sequence segment of 15000 bp centered on the TBLASTN hit and translates all the open reading frames according to GenBank annotations.
Step 5. The proteins from the highest ranking chromosome are compared to each other in order to detect potential homologs using the Smith-Waterman-Gotoh (SWG) algorithm. This procedure enables a multiple center star gene clustering topology.
Step 6. The protein sequences extracted from the highest ranking chromosome are then matched against all the proteins from the other chromosomes using the SWG algorithm. A consistent color code is assigned to matching proteins across genomes.
Step 7. Synteny maps are then drawn to scale and the corresponding open reading frames are color coded as described above.
Step 1. The protein sequence is matched against itself using BLASTP and the resulting bit score is used as the reference score (100%).
Step 2. The query protein is matched against the selected chromosomes translated in the six frames using the TBLASTN algorithm.
Step 3. The resulting scores are normalized according to the reference score determined above. Only the scores above a user-selected threshold are retained (default and minimal value of 10%). The user will also determine if only one score per chromosome or all scores are retained. The chromosomes are then ranked by decreasing scores.
Step 4. For each positive scoring chromosome, SyntTax pulls out a DNA sequence segment of 15000 bp centered on the TBLASTN hit and translates all the open reading frames according to GenBank annotations.
Step 5. The proteins from the highest ranking chromosome are compared to each other in order to detect potential homologs using the Smith-Waterman-Gotoh (SWG) algorithm. This procedure enables a multiple center star gene clustering topology.
Step 6. The protein sequences extracted from the highest ranking chromosome are then matched against all the proteins from the other chromosomes using the SWG algorithm. A consistent color code is assigned to matching proteins across genomes.
Step 7. Synteny maps are then drawn to scale and the corresponding open reading frames are color coded as described above.
| "Multiple center star" gene clustering topology |
The multiple center star gene clustering topology allows the detection of potential homologs
even in the highest ranking chromosome as shown in the following figure.
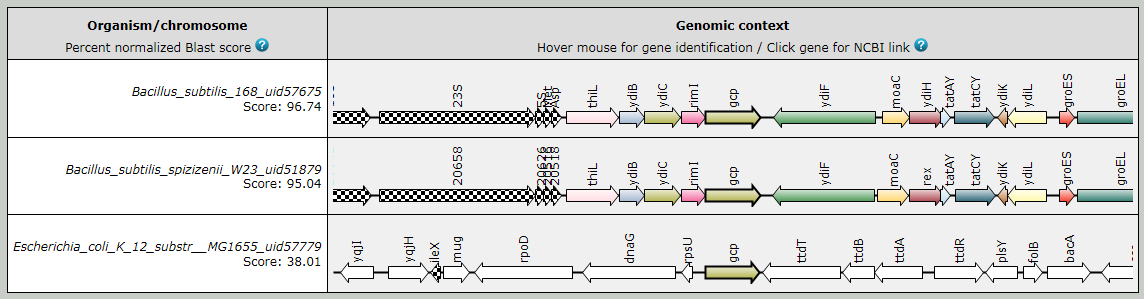
Fig.1. SyntTax example using B. subtilis 168 Gcp as query protein. In most bacteria, gcp (or ygjD) has a paralog called ydiC (or yeaZ). In many Bacillaceae, the two paralogs are in close proximity and SyntTax is able to detect these paralogs as indicated by the identical color.
Fig.1. SyntTax example using B. subtilis 168 Gcp as query protein. In most bacteria, gcp (or ygjD) has a paralog called ydiC (or yeaZ). In many Bacillaceae, the two paralogs are in close proximity and SyntTax is able to detect these paralogs as indicated by the identical color.
| Taxonomic selection tree |
The taxonomic tree for organism selection constitutes one of the most prominent properties of SyntTax.
Each completeley sequenced archaeal and bacterial organism is postioned in the tree according to its
specific lineage. The lineage is constituted of seven ranks in the following order: superkingdom, phylum,
class, family, order, genus and species. The selection of a given lineage will select all the ranks contained
in its child nodes. This intuitive selection mode according to the taxonomic classification allows
to perform
very robust synteny analyses. The user does not need to remember the classification of organisms carrying often
exotic names. Newly sequenced organims are also ranked automatically.
Taxonomic rank definitions are provided by the
NCBI Taxonomy database and are retrieved on a daily basis
by the ancillary updater program.
To my knowledge, none of the available synteny programs allows the selection
of organisms on the basis of taxonomy.
| Selected chromosomes |
A maximum number of
100 archaeal and bacterial individual chromosomes can be
selected for each search. This value has been shown
empirically to exceed the needs of typical synteny queries.
This limitation ensures also preservation of computing
resources. Upon query submission, all the chromosomes in
the listbox will be investigated.
| Insert query protein sequence |
SyntTax will accept a protein sequence in any readable format such as FASTA or raw sequence. Other formats
are accepted as well (blank spaces and numerical digits will be automatically stripped off the submitted
sequence).
| Search parameters & minimal search threshold |
To ensure an intuitive experience for the user, parameter
tweaking has been reduced to a strict minimum. The user can
determine if one score only per chromosome or all scores are
to be retained. The minimal threshold normalized BLAST bit score can be set anywhere between 10% and 100%. To obtain this score, the sequence of
the query protein is matched against itself using BLASTP and the resulting bit score is used as the
reference score of 100% for the subsequent alignments.
| Performance |
SyntTax was designed using a bottom-up approach and built with an assembly of individual components.
Each component was developped and tested separately using performance assessment. A particular interest
was devoted to the routines where most of the computing time was spent. It came as no surprise that the
optimization of the routines involved in protein alignment would produce the biggest benefits. In
particular, two solutions were instrumental in boosting SyntTax's overall performance, as follows:
1. BLAST throughtput improvement. The query protein matching against the translated genomes is carried out by running a highly optimized executable inside SyntTax (see SyntTax Workflow Step 2). The query sequence and the complete genomes are fed to the executable and its output is parsed and further processed by SyntTax. We have compared the respective performance of the BLAST and BLAT executables. The two executables are equally fast but BLAT failed in the detection of distant homologs found by BLAST. There is clearly no match for BLAST when performance is required to align a protein or a gene sequence to a complete genome. BLAST throughput was further maximized by limiting as much as possible the notoriously slow disk file read/write: query proteins sequences are fed directly from live memory (RAM) to the BLAST executable using an undocumented shell command.
2. BLAST parallelization. Several bugs have been corrected in the BLAST 2.2.26 executable now used in SyntTax. This upgrade allows full multi-threaded execution of BLAST searches by taking advantage of the multi-core architecture of modern processors. The overall speed of execution is therefore increased.
3. Use and parallelization of the Smith-Waterman-Gotoh (SWG) algorithm. Multiple protein-protein alignments are required in Step 6 of the SyntTax workflow (see above). For this particular purpose, the SWG algorithm was preferred over the most commonly used BLASTP for several reasons. i) it is a more sensitive global alignment than BLASTP, ii) it can be easily be run in parallel on multiple processor cores and iii) both the query and the subject protein sequences can be fed to SWG directly from memory without the need of disk file read/write.
1. BLAST throughtput improvement. The query protein matching against the translated genomes is carried out by running a highly optimized executable inside SyntTax (see SyntTax Workflow Step 2). The query sequence and the complete genomes are fed to the executable and its output is parsed and further processed by SyntTax. We have compared the respective performance of the BLAST and BLAT executables. The two executables are equally fast but BLAT failed in the detection of distant homologs found by BLAST. There is clearly no match for BLAST when performance is required to align a protein or a gene sequence to a complete genome. BLAST throughput was further maximized by limiting as much as possible the notoriously slow disk file read/write: query proteins sequences are fed directly from live memory (RAM) to the BLAST executable using an undocumented shell command.
2. BLAST parallelization. Several bugs have been corrected in the BLAST 2.2.26 executable now used in SyntTax. This upgrade allows full multi-threaded execution of BLAST searches by taking advantage of the multi-core architecture of modern processors. The overall speed of execution is therefore increased.
3. Use and parallelization of the Smith-Waterman-Gotoh (SWG) algorithm. Multiple protein-protein alignments are required in Step 6 of the SyntTax workflow (see above). For this particular purpose, the SWG algorithm was preferred over the most commonly used BLASTP for several reasons. i) it is a more sensitive global alignment than BLASTP, ii) it can be easily be run in parallel on multiple processor cores and iii) both the query and the subject protein sequences can be fed to SWG directly from memory without the need of disk file read/write.
| Local taxonomy database search |
The local taxonomy database can be accessed and searched for keywords or substrings. If the search is successfull,
the full lineage of the corresponding organism(s) will be returned.
| SyntTax Results |
If the search is successful, SyntTax will display a list of genomic contexts and the corresponding organisms/chromosome
names, sorted by decreasing TBLASTN scores. The gene represented in bold at the center of the synteny corrisponds to
the query protein sequence. In addition, further information can be obtained for every gene displayed in the context maps:
1. Gene identification (local). This information, obtained from the local database can be visualized by hovering the mouse cursor on any specific gene. A tooltip will pop up and indicate the full gene name, the gene product, the protein coding capacities and the specific sequence identifier number (GI).
2. Link to the NCBI database (remote). For each coding gene shown in the context maps, additional information can be retrieved by mouse clicking. A new page will open at the NCBI showing the complete protein entry constituted by the annotations and the amino acid sequence.
3. Organisms without synteny. SyntTax indicates the organims where the synteny was not found. This feature is particularly usefull when entire families of organisms are queried.
1. Gene identification (local). This information, obtained from the local database can be visualized by hovering the mouse cursor on any specific gene. A tooltip will pop up and indicate the full gene name, the gene product, the protein coding capacities and the specific sequence identifier number (GI).
2. Link to the NCBI database (remote). For each coding gene shown in the context maps, additional information can be retrieved by mouse clicking. A new page will open at the NCBI showing the complete protein entry constituted by the annotations and the amino acid sequence.
3. Organisms without synteny. SyntTax indicates the organims where the synteny was not found. This feature is particularly usefull when entire families of organisms are queried.
| Export SyntTax Acrobat .PDF Report |
This feature allows the user to save SyntTax results as a local
Acrobat .PDF file, compatible with Adobe Acrobat Reader on all operating systems
for printing, storage or further use. The Acrobat report contains all the results generated
by SyntTax. This feature allows device-independent, high-resolution printing in A4 and
Letter formats. This report is generated "on the fly" from
system memory: no user data is ever saved on the server, at
any moment.
| Export SyntTax Excel .CSV Report |
This feature allows the user to save SyntTax results as a local
Excel .CSV file, compatible with spreadsheet programs on all operating systems
for further processing. The Excel report contains most of the results generated
by SyntTax with the exception of the synteny maps. This report is generated "on the fly" from
system memory: no user data is ever saved on the server, at any moment.
| Genomic database updates |
All the SyntTax databases are stored on the server. They are constituted by genomic and taxonomic databases.
Their update is fully automated and occurs daily at 07:00 GMT/Zulu.
The genomic databases are shared with the
ABSYNTE,
BAGET and
FITBAR web services.
If your favorite genome is not found in the database, please
let us know. We would be pleased to include additional genomes (user-provided in GenBank format)
| Useful links |
•
BAGET: a web server for the effortless retrieval of prokaryotic gene context and sequence
[PubMed]
• FITBAR: a web tool for the robust prediction of prokaryotic regulons [PubMed]
• ABSYNTE: a web tool to analyze the evolution of orthologous archaeal and bacterial gene clusters [PubMed]
• National Center for Biotechnology Information (NCBI)
• NCBI Taxonomy Browser
• Smith-Waterman-Gotoh algorithm [Smith & Waterman, 1981 PubMed] [Gotoh, 1982 PubMed]
• NCBI BLAST [PubMed]
• ARCHAEA software page
• FITBAR: a web tool for the robust prediction of prokaryotic regulons [PubMed]
• ABSYNTE: a web tool to analyze the evolution of orthologous archaeal and bacterial gene clusters [PubMed]
• National Center for Biotechnology Information (NCBI)
• NCBI Taxonomy Browser
• Smith-Waterman-Gotoh algorithm [Smith & Waterman, 1981 PubMed] [Gotoh, 1982 PubMed]
• NCBI BLAST [PubMed]
• ARCHAEA software page

Help file last updated 2011 May 1st.